
| Computational Biology and Bioinformatics Laboratory |
Department Cell and Developmental Biology
Marcos J. Araúzo-Bravo, PhD
E-mail: marara (at) mpi-muenster.mpg.de; mararabra (at) yahoo.co.uk
Team
Previous members
Research subjects
In our laboratory we develop computer tools to help understand the mechanism underlying genetic regulatory networks of the toti-, pluripotential stem cells and reprogrammed cells. We are implementing an integrative approach to elucidate the cross-talk of the main pluripotency players on the molecular level, and to provide answers to the remaining questions concerning the reprogramming mechanisms. The approach is based on collecting measurements of different nature (gene expression, microRNA expression, epigenetic profiles, protein abundance, metabolic measurements) performed in a holistic way using high-throughput techniques. We combine our own measurements with data compiled from databases.
Among the topics we are currently researching are:
This page provides access to several resources related with gene regulation developed by our laboratory.
Sequence-Dependent Conformational Energy of DNA Derived
from Molecular Dynamics Simulations: Toward
Understanding the Indirect Readout Mechanism in
Protein-DNA Recognition
|
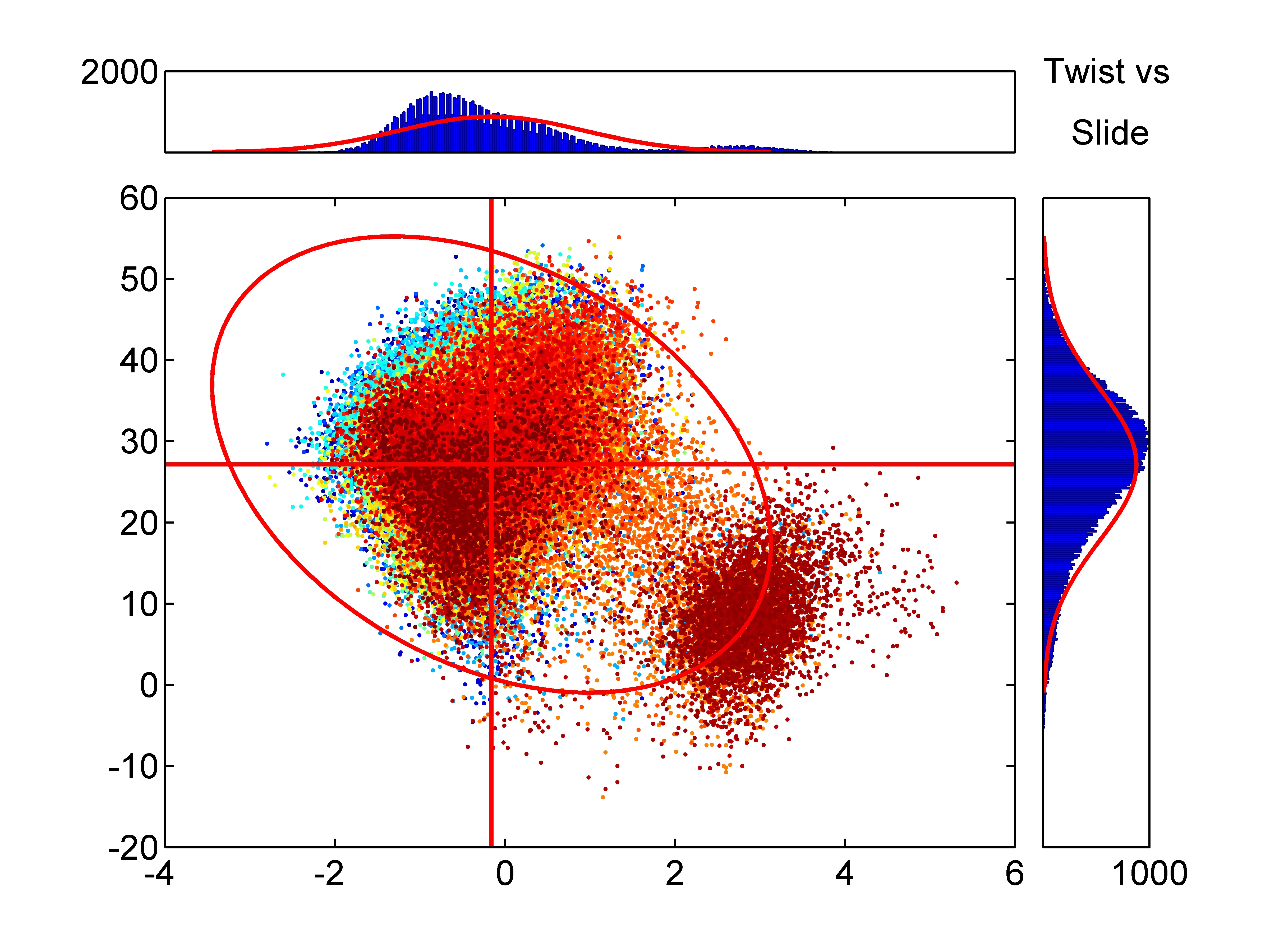
Sequence dependence of DNA conformation plays a crucial role in its recognition by proteins and ligands. To clarify the relationship between sequence and conformation, it is necessary to quantify the conformational energy and specificity of DNA. Here, we make a systematic analysis of dodecamer DNA structures including all the 136 unique tetranucleotide sequences at the center by molecular dynamics simulations. Using a simplified conformational model with six parameters to describe the geometry of adjacent base pairs and harmonic potentials along these coordinates, we estimated the equilibrium conformational parameters and the harmonic potentials of mean force for the central base-pair steps from many trajectories of the simulations. This enabled us to estimate the conformational energy and the specificity for any given DNA sequence and structure. We tested our method by using sequence-structure threading to estimate the conformational energy and the Z-score as a measure of specificity for many B-DNA and A-DNA crystal structures. The average Z-scores were negative for both kinds of structures, indicating that the potential of mean force from the simulation is capable of predicting sequence specificity for the crystal structures and that it may be used to study the sequence specificity of both types of DNA. We also estimated the positional distribution of conformational energy and Z-score within DNA and showed that they are strongly position dependent. This analysis enabled us to identify particular conformations responsible for the specificity. The presented results will provide an insight into the mechanisms of DNA sequence recognition by proteins and ligands. |
The associated article is avalaible in ( Araúzo-Bravo et al, 2005, JACS).
ReadOut: structure-based calculation of direct
and indirect readout energies and specificities for protein–DNA recognition
|
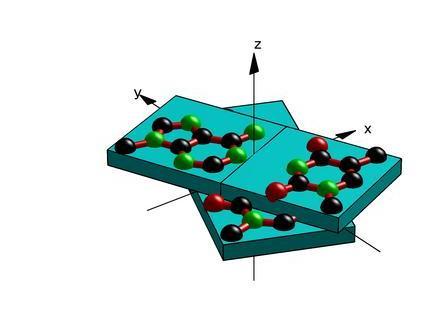
Protein–DNA interactions play a central role in regulatory processes at the genetic level. DNA-binding proteins recognize their targets by direct base–amino acid interactions and indirect conformational energy contribution from DNA deformations and elasticity. Knowledge-based approach based on the statistical analysis of protein–DNA complex structures has been successfully used to calculate interaction energies and specificities of direct and indirect readouts in protein–DNA recognition. Here, we have implemented the method as a webserver, which calculates direct and indirect readout energies and Z-scores, as a measure of specificity, using atomic coordinates of protein–DNA complexes. This server is freely available at here. The only input to this webserver is the Protein Data Bank (PDB) style coordinate data of atoms or the PDB code itself. The server returns total energy Z-scores, which estimate the degree of sequence specificity of the protein–DNA complex. This webserver is expected to be useful for estimating interaction energy and DNA conformation energy, and relative contributions to the specificity from direct and indirect readout. It may also be useful for checking the quality of protein–DNA complex structures, and for engineering proteins and target DNAs. |
The associated article is avalaible in (Ahmad et al, 2006, NAR).
Indirect readout in drug-DNA recognition: role
of sequence-dependent DNA conformation
|
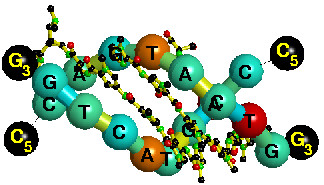
DNA-binding drugs have numerous applications in the engineered gene regulation. However, the drug-DNA recognition mechanism is poorly understood. Drugs can recognize specific DNA sequences not only through direct contacts but also indirectly through sequence-dependent conformation, in a similar manner to the indirect readout mechanism in protein-DNA recognition. We used a knowledge-based technique that takes advantage of known DNA structures to evaluate the conformational energies. We built a dataset of non-redundant free B-DNA crystal structures to calculate the distributions of adjacent base-step and base-pair conformations, and estimated the effective harmonic potentials of mean force (PMF). These PMFs were used to calculate the conforma- tional energy of drug-DNA complexes, and the Z-score as a measure of the binding specificity. Comparing the Z-scores for drug-DNA complexes with those for free DNA structures with the same sequence, we observed that in several cases the Z-scores became more negative upon drug binding. Furthermore, the specificity is position-dependent within the drug-bound region of DNA. These results suggest that DNA conformation plays an important role in the drug-DNA recognition. The presented method provides a tool for the analysis of drug-DNA recognition and can facilitate the development of drugs for targeting a specific DNA sequence. |
The associated article is avalaible in (Araúzo-Bravo and Sarai, 2008, NAR).
Comprehensive Human Transcription Factor Binding Site Map for Combinatory Binding Motifs Discovery
|
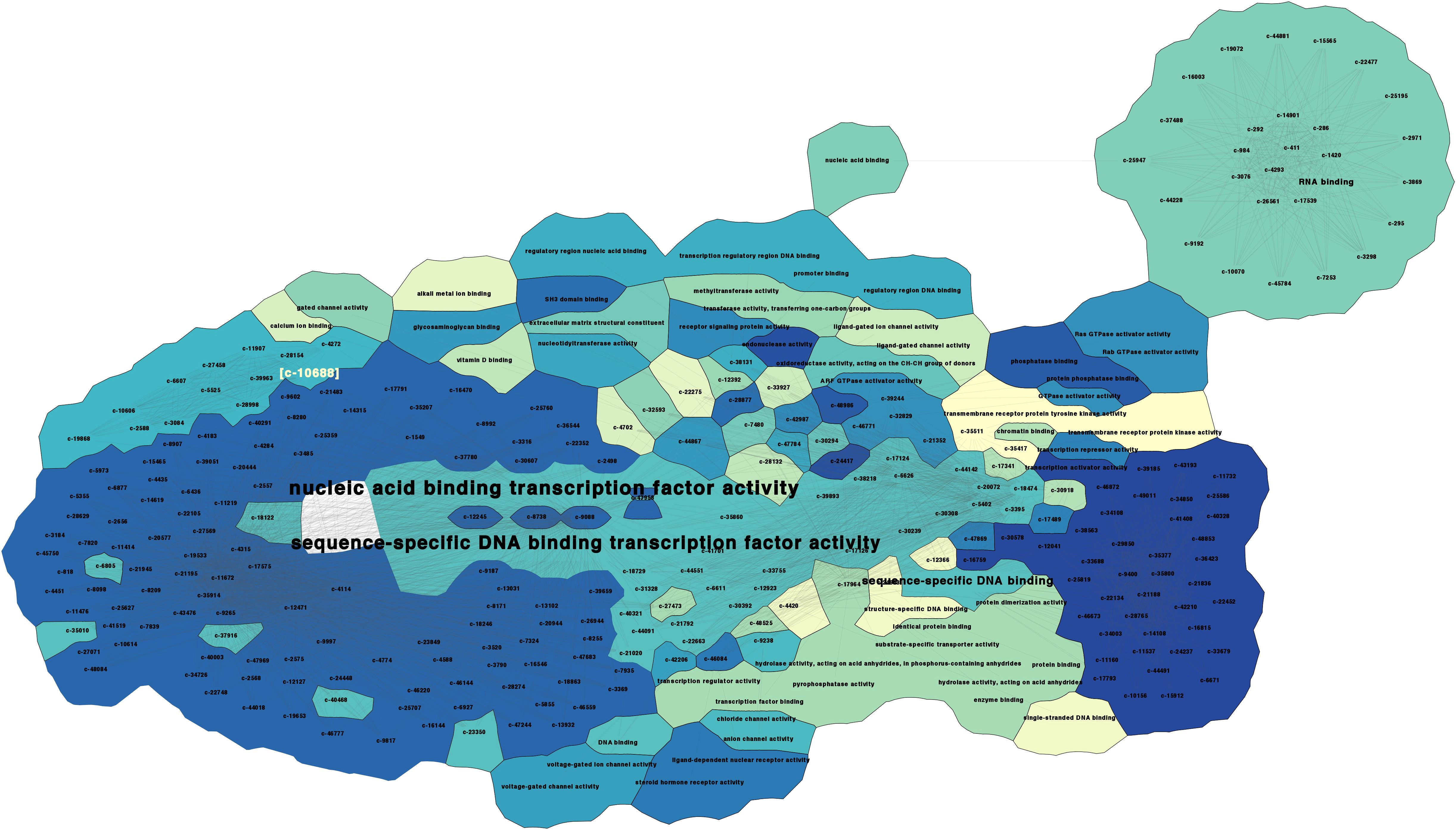
To know the map between transcription factors (TFs) and their binding sites is essential to reverse engineer the regulation process. Only about 10%–20% of the transcription factor binding motifs (TFBMs) have been reported. This lack of data hinders understanding gene regulation. To address this drawback, we propose a computational method that exploits never used TF properties to discover the missing TFBMs and their sites in all human gene promoters. The method starts by predicting a dictionary of regulatory DNA words. From this dictionary, it distills 4098 novel predictions. To disclose the crosstalk between motifs, an additional algorithm extracts TF combinatorial binding patterns creating a collection of TF regulatory syntactic rules. Using these rules, we narrowed down a list of 504 novel motifs that appear frequently in syntax patterns. We tested the predictions against 509 known motifs confirming that our system can reliably predict ab initio motifs with an accuracy of 81%—far higher than previous approaches. We found that on average, 90% of the discovered combinatorial binding patterns target at least 10 genes, suggesting that to control in an independent manner smaller gene sets, supplementary regulatory mechanisms are required. Additionally, we discovered that the new TFBMs and their combinatorial patterns convey biological meaning, targeting TFs and genes related to developmental functions. Thus, among all the possible available targets in the genome, the TFs tend to regulate other TFs and genes involved in developmental functions. We provide a comprehensive resource for regulation analysis that includes a dictionary of DNA words, newly predicted motifs and their corresponding combinatorial patterns. Combinatorial patterns are a useful filter to discover TFBMs that play a major role in orchestrating other factors and thus, are likely to lock/unlock cellular functional clusters. |
With our algorithms we generated a catalog of predictions that contains the DNA words dictionary, the motif predictions, the binding site location for each known and novel motifs, the CBPs predictions, and the visualizations for motifs and CBPs which can be downloaded from here.
The associated article is available in (Müller-Molina et al, 2012, PLoS One).
Disclosing the crosstalk among DNA methylation, transcription factors and histone marks in human pluripotent cells through discovery of DNA methylation motifs
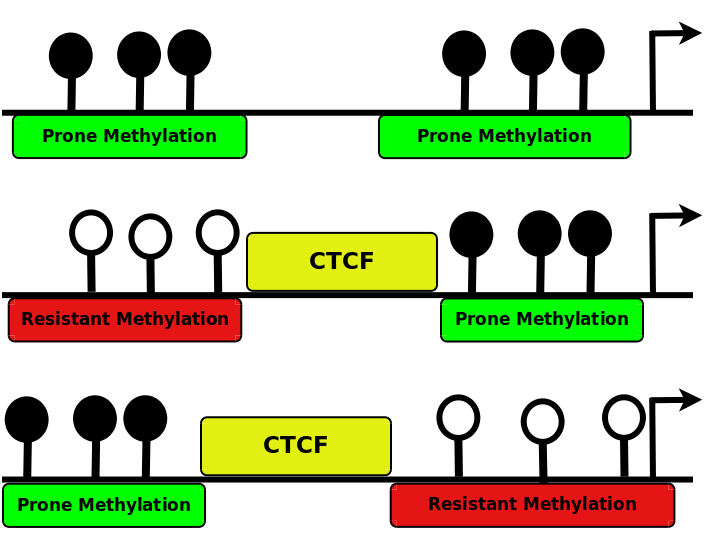
|
The known DNA methylation/unmethylation mechanisms are sequence unspecific, but different cells with the same genome have different methylomes.
Thus, additional processes bringing specificity to the methylation/unmethylation mechanisms are required.
Searching for such processes, we demonstrated that CpGs methylation states are influenced by the sequence context surrounding the CpGs.
We used such property to develop a CpG methylation motif discovery algorithm.
The newly discovered motifs reveal “methylation/unmethylation factors” that could recruit the “methylation/unmethylation machinery” to the loci specified by the motifs.
Our methylation motif discovery algorithm provides a synergistic approach to the differently methylated region algorithms.
Since our algorithm searches for commonly methylated regions inside the same sample, it requires only a single sample to operate.
The motifs found discriminate between hypomethylated and hypermethylated regions.
The hypomethylation-associated motifs have high CG content, their targets appear in conserved regions near transcription start sites,
tend to co-occur within transcription factor binding sites, are involved in breaking the H3K4me3/H3K27me3 bivalent balance,
and transit the enhancers from repressive H3K27me3 to active H3K27ac during ES cell differentiation.
The new methylation motifs characterize the pluripotent state shared between ES and iPS cells.
Additionally, we found a collection of motifs associated with the somatic memory inherited by the iPS from the initial fibroblast cells, thus revealing the existence of epigenetic somatic memory on a fine methylation scale.
With our algorithms we generated a catalog of predictions that contains the DNA methylation motifs, their gene targets, and the associated gene ontologies which can be downloaded from here. The associated article is available in (Luu et al, 2013, Genome Research). |